Всі медичні вироби класифікуються залежно від конструкції, особливостей застосування та потенційної загрози у разі їх неправильного використання. Тому перед початком процесу сертифікації, виробник медичного виробу повинен визначити його медичне призначення та передбачуване використання.
Кожна країна встановлює до них власні вимоги. Наразі в Україні діє класифікатор медичних виробів НК 024:2023, створений на основі міжнародної номенклатури GMDN.
Клас безпеки медичного виробу залежить від потенційного ризику при його застосуванні споживачем. Медичні вироби класифікують за критеріями: інвазивності, тривалості застосування, наявності контакту з тілом людини, дією на життєво важливі органи людини, а також можливості застосування джерел енергії разом з виробом.

Отже, за ступенем безпеки медичні вироби поділяються на:
Клас I — невисока доля ризику
Клас ІІа — середня доля ризику
Клас ІІб — підвищена доля ризику
Клас III — висока доля ризику
За тривалістю застосування медичні вироби бувають:
Тимчасові – для неперервного застосування до 60 хвилин;
Короткотермінові – для неперервного застосування до 30 днів;
Довготермінові – для неперервного застосування терміном від 30 днів.
За інвазивністю медичні вироби бувають:
Неінвазивні – це медичні прилади або засоби, які не потребують вторгнення чи проникнення в організм людини для проведення діагностики, моніторингу або лікування.
Інвазивні – це медичні вироби, які повністю або частково вводяться в організм людини через його поверхню або отвір тіла.
Прийняті класи безпеки медичних виробів відображають потенційний ризик у разі використання їх споживачами. Тому, залежно від визначеного класу та характеристик, процедура сертифікації медичного виробу може відрізнятись. Адже чим вищий клас ризику, тим складнішою та довшою є процедура реєстрації.
Помилки при визначенні класу можуть коштувати дорого при подальшій реалізації продукції. Тому якщо ви маєте сумніви з коректної класифікації власного виробу – звертайтесь до спеціалістів Miasphera! Ми з радістю надамо вичерпні консультації та запропонуємо оптимальні варіанти сертифікації для вашого продукту.
Залишити заявку на консультацію.
«Реєстрація косметики в Україні» – один з популярних запитів у пошукових системах. Це й не дивно, адже трендом останнього десятиліття є зростання косметичного ринку, на що вказує стабільне підвищення обсягів виробництва та реалізації косметичних засобів. Косметика давно стала не просто продуктом для догляду, а й необхідним елементом повсякденного використання людини.
Український ринок косметичних виробів не є виключенням. Тому питання появи нових продуктів, збільшення кількості виробників та технічне регулювання обігу косметики є нагальним питанням для українського законодавства.
Чи обов’язкова сертифікація косметики та коли вступає в дію новий Технічний регламент? – на ці та інші питання відповідає головний юрист компанії MIASPHERA – Віталій Ковбаса.
Перед початком опису процедури, надамо трохи теорії та розуміння терміну «косметичні засоби» з боку українського законодавства.
Косметичні засоби в Україні регулюються відповідно до Закону «Про косметичні засоби» від 05.04.2012 № 4616-VI.
Косметичною продукцією може вважатись будь-яка речовина або суміш речовин, які призначені для використання на зовнішніх ділянках тіла людини з метою їх очищення, ароматизування, відновлення, захисту та коригування запаху.
Кожен іноземний виробник чи імпортер який виводить косметичні засоби на ринок України, проходить полегшену процедуру декларування відповідності. Оскільки косметика з ЄС вже відповідає нормам, які тільки будуть впроваджені в нашій державі. Імпортеру достатньо надати сертифікат від виробника та паспорт безпеки на продукцію.
Національний виробник проходить цю процедуру через отримання висновку санітарно-епідеміологічної експертизи на виробництво та продукцію.
Наразі та до повного введення в дію Технічного регламенту, реєстрація косметичних засобів в Україні для національного виробника здійснюється через добровільне отримання висновку санітарно-гігієнічної експертизи (СЕС), який підтверджує безпеку продукції для здоров’я людини та надає право на її продаж.
Процес регулювання косметичної продукції в Україні активно інтегрується з законодавством Європейського союзу. На підставі Регламенту ЄС №1223/2009 було розроблено технічний регламент на косметичну продукцію для України. Цей нормативний акт неодноразово переносився, але все ж був прийнятий Кабміном 20 січня 2021 року.
У зв’язку з воєнними діями МОЗ запропонувало відкласти набрання чинності Технічного регламенту до 3 серпня 2024 року та запровадили перехідний період для учасників ринку косметичної продукції до 3 серпня 2026 року.

Основна мета запровадження Технічного регламенту – це встановлення нових стандартів до косметичних продуктів на ринку України, визначення прав та обов’язків учасників ринку щодо введення косметичної продукції в обіг, а також усунення адміністративних бар’єрів в торгівлі з країнами Європейського союзу.
Після набуття чинності технічного регламенту, процес реєстрації косметичної продукції стане кардинально новим.

Слід зазначити, що перехідний період, власне до моменту вступу в силу Технічного регламенту, дає час операторам ринку, щоб підготуватись. Реалізація косметики, яка виготовлена до 03.08.2026, не буде заборонена або обмежена до реалізації наступні 5 років.
Якщо ви виробник косметичних засобів та плануєте подальший розвиток власного виробництва, слід вже зараз стратегічно підходити до цього питання. Перехідний період в законодавстві – це завжди час додаткових дій та нових кроків. Щоб не втрачати час та уникнути ряду прикрих помилок при реєстрації косметичних засобів за новими правилами – звертайтесь до MIASPHERA!
Ми надамо повну юридичну підтримку та супровід на всіх етапах виводу косметичної продукції на ринок України! Фахівці MIASPHERA радо поділяться своїм успішним досвідом!
Що Miasphera може зробити для виробника косметики:
Ми завжди йдемо на крок попереду та працюємо за новими стандартами!
Бажаєте отримати консультацію? Залишайте заявку за посиланням.
Якщо ви виробник або експортер косметичної продукції та маєте бажання підкорювати ринок країн Євросоюзу – вам обов’язково необхідно пройти процедуру реєстрації ваших косметичних засобів.
Процедура проходить через оцінку відповідності косметичних виробів згідно з регламентами та директивам ЄС. Простіше кажучи: ваш продукт має відповідати стандартам ЄС на даний вид виробу.
Регламент забороняє вихід на ринок ЄС косметичних продуктів, інгредієнти або кінцевий склад яких випробовувались на тваринах. Речовини, які є канцерогенними, мутагенними чи токсичними для репродуктивного здоров’я також заборонені до використання.
Оцінка відповідності складається з проходження двох головних процедур: складання звіту з безпеки продукції (CPSR) та нотифікації. Звучить страшнувато, але в даній статті ми пояснимо це заплутане питання.
Отже, косметичні вироби охоплюють широкий спектр товарів, але все ж вони мають одну спільну рису – безпосередньо контакт з тілом людини. Саме ця особливість і є причиною ретельного вивчення фізико-хімічних показників будь-якого косметичного продукту.
Щоб впевнитися, що досліджуваний товар безпечний для використання, складається звіт з безпеки (CPSR), згідно з Додатком I Регламенту про косметику. Він містить важливі дані про результати досліджень складу та окремих компонентів, які можуть нести потенційні ризики для споживача. В процедуру також включено оформлення інформаційного файлу, де на кожен засіб зазначено відповідне маркування та умови відповідності виробництва законодавством ЄС.
Нотифікація – це процес внесення косметичної продукції в загальноєвропейську базу даних (CPNP), який проводиться на підставі звіту про безпеку продукту (про який сказано вище).
Отже, розібравшись з визначенням основних термінів, ми можемо сформувати етапи, на шляху до успішної процедури оцінки відповідності косметичних засобів:

Слід зазначити, що успішне проходження першого етапу дає змогу сформувати дорожню карту всього шляху реєстрації продукту, від якої й буде формуватися остаточний перелік необхідних документів для формування досьє – PIF файлу.
Звертаємо увагу, що у випадку реєстрації косметичної продукції на території країн Європейського Союзу, необхідно потурбуватись про наявність уповноваженого представника.
Ця відповідальна особа зареєстрована на території країни ЄС, володіє повною інформацією про продукцію, несе юридичну відповідальність за дотримання вимог по нормативах косметики та за потреби надає усю необхідну інформацію на запит наглядових органів для контролю та моніторингу діяльності.
Відповідно до Косметичного Регламенту (ЄС) No 1223/2009, уповноважений представник несе юридичну відповідальність за косметичний продукт не європейського виробника на ринку ЄС.
Всі косметичні продукти, що надходять на ринок ЄС, повинні мати наступний пакет документів:

Паспорт безпеки (MSDS) косметичної продукції розробляється на підставі даних виробника та уповноваженої особи. Він містить більш детальну інформацію про: склад, можливі ризики шкоди людині, правила поведінки в разі неправильного використання, умови зберігання та утилізації. Цей документ є важливим не лише для експортних операцій, а й для кінцевого споживача, щоб забезпечити правильне і безпечне використання товару.
У разі невідповідності косметики встановленим нормам безпеки певної країни або при поданні неповного пакета документів у реєстрації може бути відмовлено.
Бажаєте дізнатись про послугу більше?
Залишайте заявку на консультацію на сайті або телефонуйте нам! Звільніть свій час для більш важливих бізнес-процесів, а решту довірте фахівцям Miasphera! Ми працюємо по всій Україні!
Велика кількість обмежень на виробництво та вживання канабісу в різних країнах часто призводять до непорозумінь та плутанини в самих поняттях. Не всі усвідомлюють, чому існує три різні назви – канабіс, марихуана та коноплі і яка саме між ними різниця. А ті, хто не знає про існування промислових сортів, вороже ставиться до всього канабісу в цілому.
Відповідь: всі найменування вірні! Але з точки зору ботаніки, медицини та права – це абсолютно різні речі. Суть термінологічних відмінностей полягає в існуванні різних підвидів та продуктів, яки виготовлені з однієї рослини – конопель. Розберімося що і до чого.
Рослина роду Канабіс (Cannabis) або коноплі посівної – це сільськогосподарська культура, яка характеризується зазубреним листям та має характерний запах.
Ботаніки розділяють ці рослини за будовою та класифікують на основі морфології, фізіології та походження. Найбільш відомими підвидами є Sativa та Indica або їх гібриди.
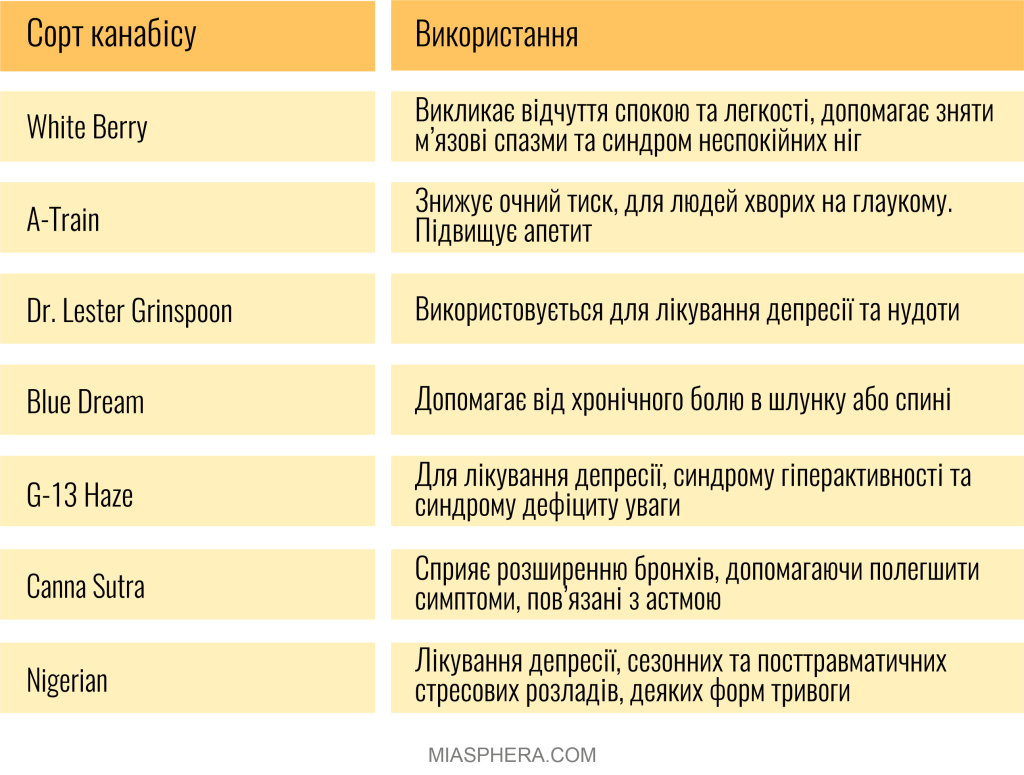
Сатива (Sativa) – це висока рослина з тонкими листям, яка традиційно вирощується в країнах Латинської Америки, Північної Африки та Південно-Східної Азії. Підвид має підвищений рівень тетрагідроканабінолу (ТГК).
Індіка (Indica) – низькорослий підвид, який відрізняється підвищеним рівнем канабідіолу (КБД) та вирощується переважно у гірських районах Індії та Пакистану. Існує також дикий підвид – Рудераліс (Ruderalis), він є менш поширеним, але на його базі були виведені всі культурні сорти технічної коноплі з майже нульовим вмістом тетрагідроканабінолу (ТГК). Він відомий своєю особливістю автоцвітіння, в залежності від віку, а не світлових циклів. Через низький вміст ТГК він не використовується в рекреаційних або медичних цілях. В деяких країнах цей промисловий вид конопель називають хемпом (hemp).
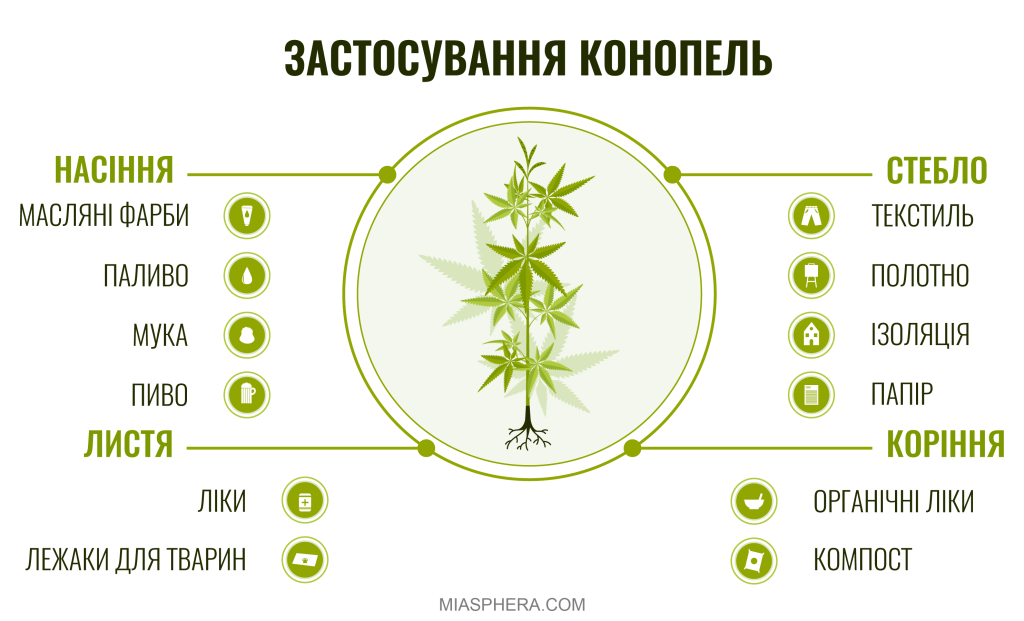
Загалом, канабіс – це латинське найменування рослини, яким також позначають медичні сорти коноплі та продукцію на основі конопель терапевтичного призначення. Традиційно, медичний канабіс відрізняється збалансованим вмістом КБД та низьким рівнем ТГК.
Марихуана – це курильна суміш із висушених суцвіть жіночих особин сативи та індики. Марихуана має більший відсоток вмісту психоактивних речовин (ТГК більше ніж 0.3%), у порівнянні з коноплями та має психотропні властивості (легкого наркотику).
У зв’язку з легалізацією медичного канабісу виникла необхідність називати різні підвиди конопель та продуктів на їх основі. Таким чином, виробники дистанціювали тих, хто потребував використання цих рослин саме в лікувальних цілях від користувачів рекреаційного споживання.
З розвитком легального ринку продуктів на основі канабісу, слід чітко розмежовувати ці найменування. Отже, знаючи властивості, можна резюмувати що:
Коноплі – це сільськогосподарська культура, яка охоплює всі сорти Cannabis ruderalis. Коноплі є джерелом екологічного текстилю та паперу, широко використовується у промислових цілях.
Канабіс – позначення спеціалізованих сортів, виведених для терапевтичних цілей, незалежно від їх наркотичних властивостей.
Марихуана – це курильна суміш із висушених суцвіть жіночих особин сативи та індики, що має властивості легкого наркотику.
Медична марихуана або медичний канабіс — цей термін використовується саме для похідних канабісу Sativa, які використовуються для полегшення симптомів, викликаних певними захворюваннями.
Основна відмінність – це у вміст ТГК. Прирівнювати медичну марихуану до рекреаційної некоректно, оскільки її завдання – не приносити задоволення, а полегшувати больові симптоми. А отже медичний канабіс – це спеціально виведені сорти зі зміненим вмістом ТГК.
Наразі виведено безліч сортів рослин канабісу Sativa саме для медичного застосування.
Основною дієвою речовиною є канабідіол (КБД). Результати наукових досліджень показують, що рослинні препарати з КБД ефективно блокують різні типи болю, підвищують апетит, допомагають подолати депресивні стани, стримують напади епілепсії. Це важливий засіб для людей, які страждають від постійних болів, онкології та СНІДу.
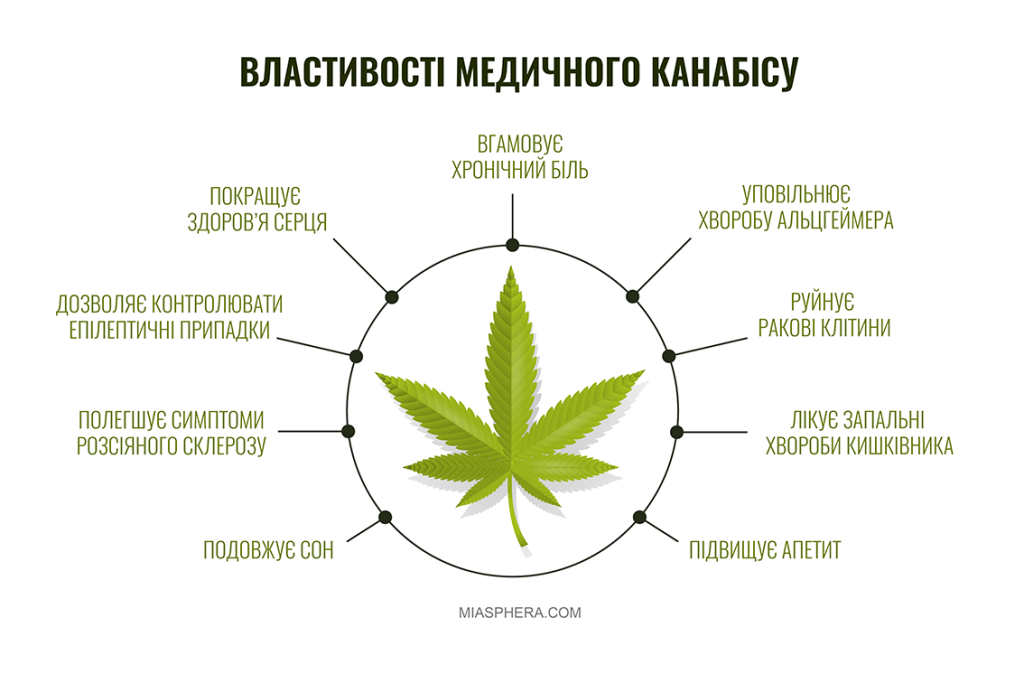
Медичний канабіс продемонстрував потенціал у полегшенні симптомів, пов’язаних з неврологічними захворюваннями, такими як епілепсія, розсіяний склероз, хвороба Паркінсона, хвороба Альцгеймера та невропатичний біль. Але важливо розуміти, що канабіс не панацея і таке лікування може бути корисним не для всіх.
Наразі у світі проводиться велика кількість досліджень на тему впливу медичного канабісу при лікуванні особливо складних хвороб.
Але водночас, неоднозначний юридичний статус різних хімотипів ускладнює вивчення властивостей канабісу. Обов’язковою умовою проведення досліджень є спеціальна ліцензія, отримання якої є досить бюрократичною процедурою.
Маємо надію, що наука у цьому питання буде уперто рухатись вперед, відкриваючи нові корисні способи застосування конопель та їх похідних в медицині, хімічній промисловості, промисловості та сільському господарстві.
Увага! Стаття розміщена з пізнавальною метою і не пропагує вживання наркотиків. Сподіваємось наша публікація принесла наукове розуміння теми та допомогла розібратись з відмінностями термінів.
Наразі без сумніву кожен світовий виробник медичних виробів вирішує для себе складне питання: «Чи імпортувати медичні вироби в Україну?» І відповідь на нього дуже проста: «Так, звісно».
Для багатьох світових виробників медичних виробів, які були присутні на ринку України до війни, наразі при діючих дозвільних документах на медичні вироби настав зірковий час, час коли Україна поступово відбудовує та відновлює пошкоджену медичну інфраструктуру.

Державним бюджетом України на 2022 рік на охорону здоров’я у бюджеті закладено 195,9 млрд грн, а з додатковим фінансуванням через повномасштабну війну – 232,8 млрд грн. В 2023 році на охорону здоров’я у бюджеті закладено 175 млрд грн, в тому числі бюджет на централізовані закупівлі медичних виробів за 31 напрямом у 2023 році становить 10,3 млрд грн.
Проте, Україна бере курс на стандартизацію до Європи, та під час відновлення та відбудови втраченої медичної інфраструктури націлена на впровадження новітніх технологій та продуктів, зокрема медичних виробів.
Тому, саме час світовим виробникам, що мають новітні та передові медичні технології та продукти відкрити для себе ринок України та почати здійснювати імпорт медичних виробів в Україну.
Подолати всі складності пов’язані з імпортом медичних виробів в Україну світовому виробнику медичних виробів допоможе призначений в Україні Уповноважений представник. Оскільки бідь-який імпорт медичних виробів в Україну для виробників медичних виробів, які не є резидентами України, не можливий без призначення Уповноваженого представника в Україні.
Саме Уповноважений представник, що може бути учасником торгівлі так і виключно тримачем реєстраційних документів на медичний виріб, здійснює всі необхідні дії щоб імпорт медичних виробів в Україну був успішний.
Для імпорту необхідно здійснити ряд обов’язкових дій так і додаткових дій на вибір виробника медичних виробів. Обов’язково необхідно укласти договір з компанією з України про надання послуг Уповноваженого представника.
Маючи підтвердження своїх повноважень, Уповноважений представник вивчає технічну документацію на медичний виріб, з метою встановлення необхідної процедури оцінки відповідності медичного виробу, та в подальшому ініціює її проведення. Тобто, імпорт медичних виробів в Україну, що відносяться до медичних виробів I-го класу, нестерильних, без функції вимірювання та медичних виробів для діагностики in vitro, і не призначені для самоконтролю можливий при реєстрації виробника та Уповноваженого представника до Реєстру Державної служби України з лікарських засобів та контролю за наркотиками. Натомість, медичні вироби більш високого класу ризику підлягають проходженню процедури аудиту виробника із залученням призначеного органу з оцінки відповідності медичних виробів.
Маючи такі документи як декларація про відповідність медичних виробів та/або сертифікат про оцінку відповідності на медичний виріб – найскладніші етапи перед здійсненням імпорту пройдені.
Законодавство України дозволяє здійснювати імпорт медичних виробів в Україну за умови підтвердження їх відповідності Технічним регламентам (що підтверджується згаданими вище декларацією та/або сертифікатом), розробленими у відповідності до вимог українською мовою етикетки на медичний виріб, інструкція використання медичного виробу.
У зв’язку з тим, що в Україні як і в інших країнах, обіг медичних виробів регульований, до згаданих етикеток та інструкції на медичний виріб встановлюється ряд вимог до їх форми та змісту. І цю складну задачу бере на себе Уповноважений представник, від виробника медичних виробів потребуватиметься лише надання технічних файлів на медичний виріб, опрацювавши, які Уповноважений представник розробить вірну етикетку та інструкцію на медичний вирі.
Зазначені вище документи підлягають перевірці на митниці під час проходження регламентованих процедур розмитнення медичних виробів на території України, які визначаються в залежності від медичного виробу, згідно до коду УКТ ЗЕД на виріб.
З цього етапу, виробник медичних виробів може здійснювати виробництво таких виробів щоб здійснити імпорт медичних виробів в Україну. І користь Уповноваженого представника на наступних етапах є не менш важлива, оскільки Уповноважений представник може здійснити пошук логістичного партнера для імпорту товарів, порадити надійного дилера, здійснити дії направлені на запобігання сірого імпорту медичних виробів виробника на ринку України. Перелічені дії є додатковими для виробника медичних виробів, але не менш важливими для успішного не тільки імпорту, але й реалізації медичних виробів на ринку України.
Імпорт медичних виробів в Україну є простим, не дорогим та передбачувано успішним, якщо доручити цю справу надійному партнеру – Уповноваженому представнику. Імпортуйте медичні вироби в Україну – долучайтеся до відбудови та відновлення України, за мир, справедливість і демократичні цінності.
В Україні ще з часу запровадження дій направлених на запобігання поширенню коронавірусної інфекції SARS-CoV-2, а саме з березня 2020 року, запроваджено можливість проведення процедури оцінки відповідності медичних виробів шляхом дистанційного аудиту виробника. Підтвердженням завершення проходження зазначеної процедури є сертифікат відповідності. В Україні, як і в решті країн світу, введення в обіг та реалізація медичних виробів можлива за умови підтвердження їх відповідності технічним регламентам на медичні вироби, тобто отримання на медичні вироби сертифікату відповідності та/або декларації відповідності.
Нажаль, з 24 лютого 2022 року від дня військової агресії рф проти України, та у зв’язку з продовженням карантинних обмежень через коронавірусну інфекцію SARS-CoV-2, в Україні зберіглася можливість проведення процедури оцінки відповідності медичних виробів шляхом дистанційного аудиту виробника.
Щоб отримати сертифікат відповідності під час воєнного стану в Україні, виробник медичних виробів, який не є резидентом України, через свого Уповноваженого представника здійснює звернення до призначеного органу з оцінки відповідності та ініціює процес оцінки відповідності медичних виробів шляхом підписання договору.
Наразі, всі призначені органи з оцінки відповідності в Україні забезпечують можливість проходження процедури оцінки відповідності медичних виробів та по завершенню видають сертифікат відповідності під час воєнного стану в Україні.
У зв’язку з можливістю дистанційного аудиту виробника в умовах воєнного стану ця процедура передбачає собою оцінку документів та записів, що здійснюється шляхом використання віддаленого доступу.
– високу оперативність проведення робіт з оцінки відповідності медичних виробів;
– можливість залучати до перевірки важкодоступний персонал в усьому світі і кількох країнах одночасно;
– скорочення витрат на переїзд та сполучення;
– зменшення робочого часу співробітників призначеного органу з оцінки відповідності.
В загальному підсумку зниження вартості процедури з оцінки відповідності шляхом аудиту виробника є суттєвою перевагою. До зазначеної переваги швидкості дистанційного аудиту та меншої вартості такої процедури в порівнянні з виїзним аудитом додається і те, що наразі медичний ринок України потребує значної кількості нових медичних виробів, серед яких загальновживаних додалися нові – тактична медицина, вироби для реабілітації та протезування.
В Україні через війну з рф, відповідно до дослідження Світового банку станом на 2023 рік через, зруйновано або пошкоджено щонайменше 978 медичних закладів. Також пошкоджено або знищено 650 «швидких» та щонайменше 596 аптек. Проте, не дивлячись на те, що війна продовжується, уряд України поводить курс на відновлення втраченої медичної інфраструктури, а тому медичний ринок потребує як ніколи великих обсягів медичних товарів від бинтів до медичного обладнання.
Зокрема ринок лабораторних досліджень на прикладі систем для забору крові представлений в Україні 428 центрами, де здають і зберігають кров, з них 43 центри крові обласного та міського значення; 309 відділень трансфузіології; 76 лікарень де приймають донорів. Зазначена медична інфраструктура потребує значних обсягів різних витратних матеріалів, а також пробірок вакуумних для забору крові, голок для забору крові, контейнерів для транспортування та зберігання крові, дезінфікуючих засобів, рукавичок та одягу для медичного персоналу тощо.
Отже, сертифікат відповідності під час воєнного стану в України продовжується видаватися призначеними органами з оцінки відповідності, і є результатом процедури оцінки відповідності медичних виробів шляхом дистанційного аудиту виробника.
Реєстрація виробів медичного призначення в Україні як і в інших країнах світу передбачає собою їх легалізацію та необхідна для введення виробником в обіг виробів медичного призначення.
З 2015 року в Україні змінено процедуру державної реєстрації медичних виробів і впроваджено процедуру оцінки відповідності вимогам технічних регламентів щодо медичних виробів, у тому числі для діагностики in vitro та активних медичних виробів, які імплантують, що передбачено Законом України «Про технічні регламенти та оцінку відповідності».
Найважливіше про що варто одразу зазначити, на даний час в Україні не скасовані карантинні заходи направленні на запобігання та попередження поширення COVID-19, та на превеликий жаль через військову агресію (війну) рф проти України, процедура оцінки відповідності вимогам технічних регламентів щодо медичних виробів із залученням призначеного органу тимчасово відбувається дистанційно. Дистанційний аудит виробника медичних виробів має такі переваг як: швидкість аудиту та вартість такого аудиту. Виробнику виробів медичного призначення не потрібно оплачувати переліт аудиторів та перекладача з України в країну виробництва виробів медичного призначення, оплачувати кошти на проживання в готелі, добові витрати тощо.
Відповідно реєстрація виробів медичного призначення в Україні здійснюється згідно до порядку, визначеного Технічним регламентом, що залежить типу та класу ризику виробу медичного призначення.
Перший спосіб є найпростішим та передбачає завершення внутрішнього контролю оформленням та підписанням під цілковиту відповідальність виробника декларації про відповідність виробів медичного призначення вимогам технічних регламентів. По результату внутрішнього контролю, уповноважений представник, отримані документи від виробника на вироби медичного призначення – технічний файл, розроблені етикетки, інструкції зберігає не тільки на час реалізації виробів медичного призначення, але й до 15 років після реалізації останнього виробу. Після оформлення декларації, отримання документів на вироби медичного призначення, уповноважений представник повинен здійснити внесення запису до Реєстру осіб відповідальних за введення медичних виробів у обіг. Від дня внесення такого запису, виробник та/або його уповноважений представник мають право здійснювати імпортування виробів медичного призначення в Україну за пільговою ставкою податку на додану вартість та здійснювати реалізацію таких виробів на ринку України.
В загальному порядку, другий спосіб є більш складним та тривалішою процедурою, що поділяється на декілька етапів: звернення до органу з оцінки відповідності з відповідною заявою, до якої додаються анкетні форми, списки виробів медичного призначення; проходження експертизи документів на вироби медичного призначення – технічного файлу; аудит системи управління якістю виробника. За результатом аудиту призначений орган з оцінки відповідності видає сертифікат про відповідність. Після отримання сертифікату про відповідність, виробник та/або уповноважений представник здійснює оформленням та підписанням під цілковиту відповідальність виробника декларації про відповідність виробів медичного призначення вимогам технічних регламентів.
На даний час існують всі передумови для того щоб реєстрація виробів медичного призначення в Україні була вигідною для виробника, коштувала в рази дешевше та проводилася швидше. Зрештою, саме час для світових виробників виробів медичного призначення відкрити для себе медичний ринок України, та посісти в ньому гідне місце. Реєстрація виробів медичного призначення в Україні, які через відновлення критичної медичної інфраструктури є вкрай необхідні по всім медичним направленням, надасть світовому виробнику перевагу в освоєнні медичного ринку України.







